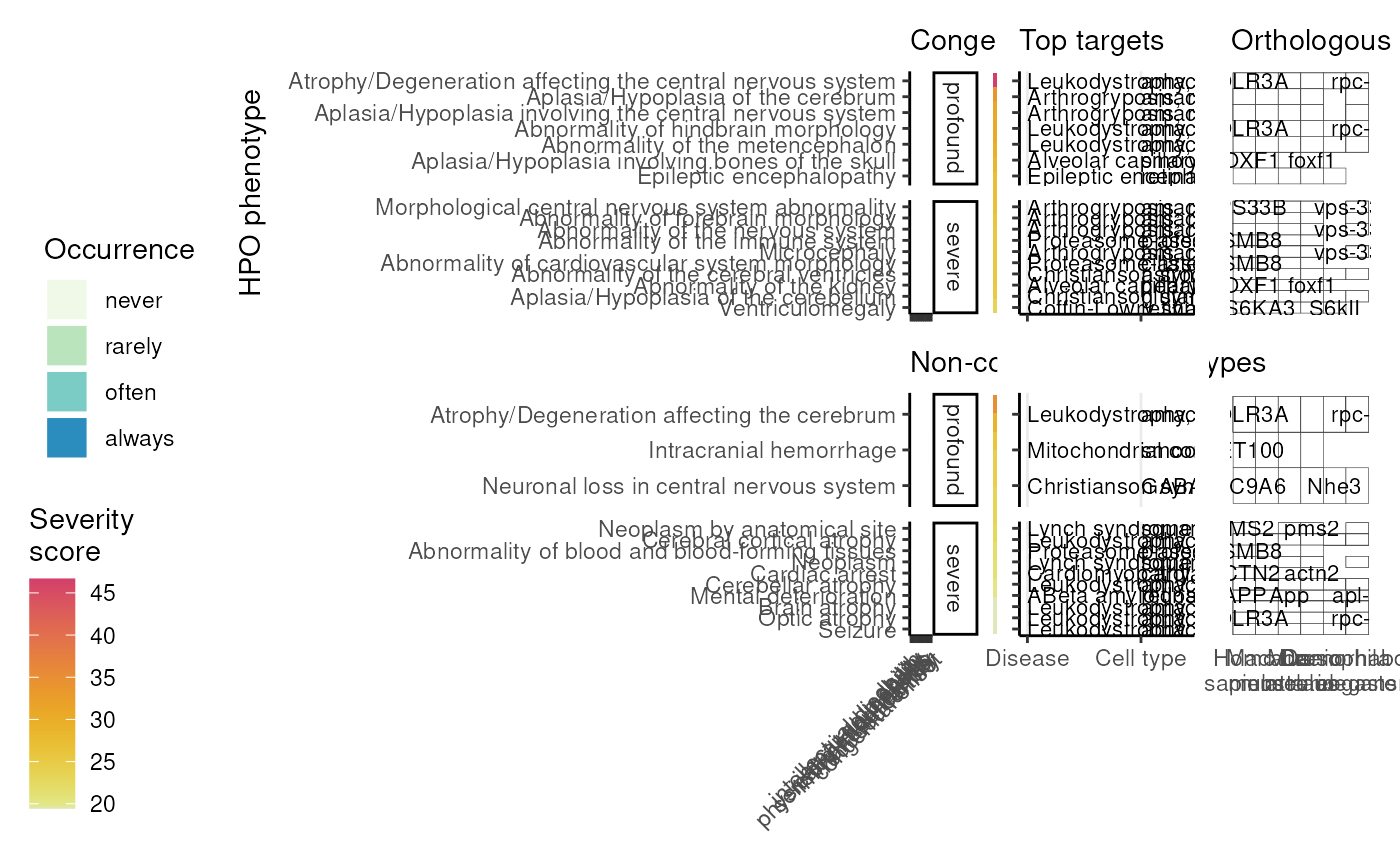
Plot the output of prioritise_targets as a grid.
prioritise_targets_grid(
top_targets,
res_class = HPOExplorer::gpt_annot_class(),
n_per_class = 10,
keep_severity_class = c("profound", "severe"),
keep_physical_malformations = NULL,
species_list = c("Homo sapiens", "Macaca mulatta", "Mus musculus", "Danio rerio",
"Drosophila melanogaster", "Caenorhabditis elegans"),
legend.position = "left",
keep_ont_levels = NULL,
width = 70,
widths1 = c(1, 4),
widths2 = c(1, 0.8),
show_plot = TRUE,
...
)Arguments
- top_targets
output of prioritise_targets.
- res_class
Output of the gpt_annot_class function.
- n_per_class
Number of phenotypes per severity class to include.
- keep_severity_class
Phenotypes to keep based on severity classes.
- keep_physical_malformations
Phenotypes to keep based on physical malformation frequency (0=never, 1=rarely, 2=often, 3=always).
- species_list
Species to include in orthologous genes grid.
- legend.position
the default position of legends ("none", "left", "right", "bottom", "top", "inside")
- keep_ont_levels
Only keep phenotypes at certain absolute ontology levels to keep. See add_ont_lvl for details.
- width
Width of the saved plot.
- widths1
Proportional widths of severity annotation heatmap (left subplot) and the top targets/orthologous genes grid (right subplot).
- widths2
Proportional widths of the top targets/orthologous genes grid.
- show_plot
Print the plot to the console.
- ...
Arguments passed on to
HPOExplorer::plot_top_phenoskeep_descendantsTerms whose descendants should be kept (including themselves). Set to
NULL(default) to skip this filtering step.annotation_orderThe order of the annotations to include.
split_by_congenitalSplit the phenotypes by congenital onset (congenital = always/often, noncongenital = never/rarely).
axis.text.xWhether to include x-axis text in top and bottom subplots.
Value
A named list with data and patchwork object.
Examples
top_targets <- MSTExplorer::example_targets$top_targets
out <- prioritise_targets_grid(top_targets = top_targets)
#> Mapping cell types to cell ontology terms.
#> Adding stage information.
#> Adding disease_name and disease_description.
#> Translating ontology terms to ids.
#> Reading cached RDS file: phenotype_to_genes.txt
#> + Version: v2025-11-24
#> 151 phenotypes do not have matching HPO IDs.
#> Reading in GPT annotations for 16,982 phenotypes.
#> Assigning severity classes.
#> Adding level-2 ancestor to each HPO ID.
#> Adding ancestor metadata.
#> Ancestor metadata already present. Use force_new=TRUE to overwrite.
#> Translating ontology terms to ids.
#> Keeping descendants of 1 term(s).
#> 17,548 terms remain after filtering.
#> 1,150 associations remain after filtering.
#> Getting absolute ontology level for 18,082 IDs.
#> Adding disease_name and disease_description.
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 17 genes extracted.
#> Converting human ==> Macaca mulatta orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Macaca mulatta
#> 1 organism identified from search: 9544
#> Checking for genes without orthologs in Macaca mulatta.
#> Extracting genes from input_gene.
#> 7 genes extracted.
#> Extracting genes from ortholog_gene.
#> 7 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 0 / 7 (0%)
#> Total genes remaining after convert_orthologs :
#> 7 / 7 (100%)
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 17 genes extracted.
#> Converting human ==> Mus musculus orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Mus musculus
#> 1 organism identified from search: 10090
#> Checking for genes without orthologs in Mus musculus.
#> Extracting genes from input_gene.
#> 7 genes extracted.
#> Extracting genes from ortholog_gene.
#> 7 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 0 / 7 (0%)
#> Total genes remaining after convert_orthologs :
#> 7 / 7 (100%)
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 17 genes extracted.
#> Converting human ==> Danio rerio orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Danio rerio
#> 1 organism identified from search: 7955
#> Checking for genes without orthologs in Danio rerio.
#> Extracting genes from input_gene.
#> 7 genes extracted.
#> Extracting genes from ortholog_gene.
#> 7 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 0 / 7 (0%)
#> Total genes remaining after convert_orthologs :
#> 7 / 7 (100%)
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 17 genes extracted.
#> Converting human ==> Drosophila melanogaster orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Drosophila melanogaster
#> 1 organism identified from search: 7227
#> Checking for genes without orthologs in Drosophila melanogaster.
#> Extracting genes from input_gene.
#> 4 genes extracted.
#> Extracting genes from ortholog_gene.
#> 4 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 3 / 7 (43%)
#> Total genes remaining after convert_orthologs :
#> 4 / 7 (57%)
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 17 genes extracted.
#> Converting human ==> Caenorhabditis elegans orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Caenorhabditis elegans
#> 1 organism identified from search: 6239
#> Checking for genes without orthologs in Caenorhabditis elegans.
#> Extracting genes from input_gene.
#> 5 genes extracted.
#> Extracting genes from ortholog_gene.
#> 5 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Dropping 1 genes that have multiple input_gene per ortholog_gene (many:1).
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 4 / 7 (57%)
#> Total genes remaining after convert_orthologs :
#> 3 / 7 (43%)
#> Adding disease_name and disease_description.
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 13 genes extracted.
#> Converting human ==> Macaca mulatta orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Macaca mulatta
#> 1 organism identified from search: 9544
#> Checking for genes without orthologs in Macaca mulatta.
#> Extracting genes from input_gene.
#> 6 genes extracted.
#> Extracting genes from ortholog_gene.
#> 6 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 1 / 7 (14%)
#> Total genes remaining after convert_orthologs :
#> 6 / 7 (86%)
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 13 genes extracted.
#> Converting human ==> Mus musculus orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Mus musculus
#> 1 organism identified from search: 10090
#> Checking for genes without orthologs in Mus musculus.
#> Extracting genes from input_gene.
#> 7 genes extracted.
#> Extracting genes from ortholog_gene.
#> 7 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 0 / 7 (0%)
#> Total genes remaining after convert_orthologs :
#> 7 / 7 (100%)
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 13 genes extracted.
#> Converting human ==> Danio rerio orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Danio rerio
#> 1 organism identified from search: 7955
#> Checking for genes without orthologs in Danio rerio.
#> Extracting genes from input_gene.
#> 7 genes extracted.
#> Extracting genes from ortholog_gene.
#> 7 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 0 / 7 (0%)
#> Total genes remaining after convert_orthologs :
#> 7 / 7 (100%)
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 13 genes extracted.
#> Converting human ==> Drosophila melanogaster orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Drosophila melanogaster
#> 1 organism identified from search: 7227
#> Checking for genes without orthologs in Drosophila melanogaster.
#> Extracting genes from input_gene.
#> 2 genes extracted.
#> Extracting genes from ortholog_gene.
#> 2 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 5 / 7 (71%)
#> Total genes remaining after convert_orthologs :
#> 2 / 7 (29%)
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from value.
#> 13 genes extracted.
#> Converting human ==> Caenorhabditis elegans orthologs using: homologene
#> Retrieving all organisms available in homologene.
#> Mapping species name: human
#> Common name mapping found for human
#> 1 organism identified from search: 9606
#> Retrieving all organisms available in homologene.
#> Mapping species name: Caenorhabditis elegans
#> 1 organism identified from search: 6239
#> Checking for genes without orthologs in Caenorhabditis elegans.
#> Extracting genes from input_gene.
#> 4 genes extracted.
#> Extracting genes from ortholog_gene.
#> 4 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 3 / 7 (43%)
#> Total genes remaining after convert_orthologs :
#> 4 / 7 (57%)