Automatically creates a phylogenetic tree plot annotated with metadata
describing how many orthologous genes each species shares with the
reference_species ("human" by default).
plot_orthotree(
tree = NULL,
orth_report = NULL,
species = NULL,
method = c("babelgene", "homologene", "gprofiler"),
tree_source = "timetree",
non121_strategy = "drop_both_species",
reference_species = "human",
clades = list(Primates = c("Homo sapiens", "Macaca mulatta"), Eutherians =
c("Homo sapiens", "Mus musculus", "Bos taurus"), Mammals = c("Homo sapiens",
"Mus musculus", "Bos taurus", "Ornithorhynchus anatinus", "Monodelphis domestica"),
Tetrapods = c("Homo sapiens", "Mus musculus", "Gallus gallus", "Anolis carolinensis",
"Xenopus tropicalis"), Vertebrates = c("Homo sapiens", "Mus musculus",
"Gallus gallus", "Anolis carolinensis", "Xenopus tropicalis", "Danio rerio"),
Invertebrates = c("Drosophila melanogaster",
"Caenorhabditis elegans")),
clades_rotate = list(),
scaling_factor = NULL,
show_plot = TRUE,
save_paths = c(tempfile(fileext = ".ggtree.pdf"), tempfile(fileext = ".ggtree.png")),
width = 15,
height = width,
mc.cores = 1,
verbose = TRUE
)Source
Arguments
- tree
A phylogenetic tree of class phylo. If no tree is provided (
NULL) a 100-way multiz tree will be imported from UCSC Genome Browser.- orth_report
An ortholog report from one or more species generated by report_orthologs.
- species
Species to include in the final plot. If
NULL, then all species from the given database (method) will be included (via map_species), so long as they also exist in thetree.- method
R package to use for gene mapping:
"gprofiler"Slower but more species and genes.
"homologene"Faster but fewer species and genes.
"babelgene"Faster but fewer species and genes. Also gives consensus scores for each gene mapping based on a several different data sources.
- tree_source
Can be one of the following:
- "timetree2022":
Import and prune the TimeTree >147k species phylogenetic tree. Can also simply type "timetree".
- "timetree2015":
Import and prune the TimeTree >50k species phylogenetic tree.
- "OmaDB":
Construct a tree from OMA (Orthologous Matrix browser) via the getTaxonomy function. NOTE: Does not contain branch lengths, and therefore may have limited utility.
- "UCSC":
Import and prune the UCSC 100-way alignment phylogenetic tree (hg38 version).
- "<path>":
Read a tree from a newick text file from a local or remote URL using read.tree.
- "timetree2022":
- non121_strategy
How to handle genes that don't have 1:1 mappings between
input_species:output_species. Options include:"drop_both_species" or "dbs" or 1Drop genes that have duplicate mappings in either the
input_speciesoroutput_species(DEFAULT)."drop_input_species" or "dis" or 2Only drop genes that have duplicate mappings in the
input_species."drop_output_species" or "dos" or 3Only drop genes that have duplicate mappings in the
output_species."keep_both_species" or "kbs" or 4Keep all genes regardless of whether they have duplicate mappings in either species.
"keep_popular" or "kp" or 5Return only the most "popular" interspecies ortholog mappings. This procedure tends to yield a greater number of returned genes but at the cost of many of them not being true biological 1:1 orthologs.
"sum","mean","median","min" or "max"When
gene_dfis a matrix andgene_output="rownames", these options will aggregate many-to-one gene mappings (input_species-to-output_species) after dropping any duplicate genes in theoutput_species.
- reference_species
Reference species.
- clades
A named list of clades each containing a character vector of species used to define the respective clade using MRCA.
- clades_rotate
A list of clades to rotate (via rotate), each containing a character vector of species used to define the respective clade using MRCA.
- scaling_factor
How much to scale y-axis parameters (e.g. offset) by.
- show_plot
Whether to print the final tree plot.
- save_paths
Paths to save plot to.
- width
Saved plot width.
- height
Saved plot height.
- mc.cores
Number of cores to parallelise different steps with.
- verbose
Print messages.
Value
A list containing:
- plot :
Annotated ggtree object.
- tree :
The pruned, standardised phylogenetic tree used in the plot.
- orth_report :
Ortholog reports for each
speciesagainst thereference_species
.
- metadata :
Metadata used in the plot, including silhouette PNG ids from phylopic.
- clades :
Metadata used for highlighting clades.
- method :
methodused.- reference_species :
reference_speciesused.- save_paths :
save_pathsto plot.
Examples
if(require("ape")){
suppressWarnings(
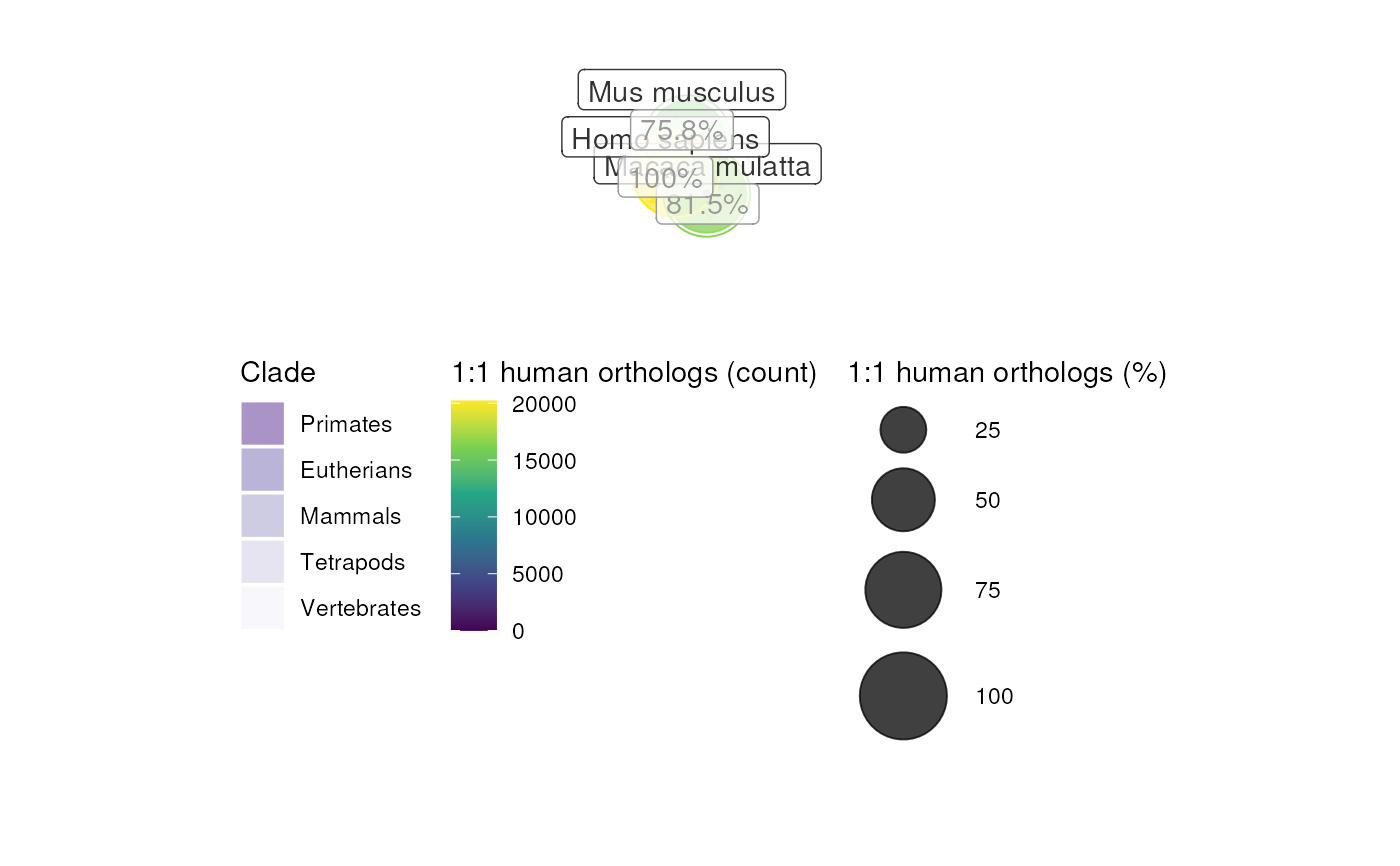
orthotree <- plot_orthotree(species = c("human","monkey","mouse"))
)
}
#> Loading required package: ape
#> Gathering ortholog reports.
#> Retrieving all genes using: babelgene.
#> Retrieving all organisms available in babelgene.
#> Mapping species name: Homo sapiens
#> 1 organism identified from search: 9606
#> Using cached file: /github/home/.cache/R/orthogene/all_genes-9606-babelgene.csv.gz
#> Returning all 20,492 genes from Homo sapiens.
#> Retrieving all genes using: babelgene.
#> Retrieving all organisms available in babelgene.
#> Mapping species name: Homo sapiens
#> 1 organism identified from search: 9606
#> Using cached file: /github/home/.cache/R/orthogene/all_genes-9606-babelgene.csv.gz
#> Returning all 20,492 genes from Homo sapiens.
#> --
#>
#> =========== REPORT SUMMARY ===========
#> 20,206 / 20,206 (100%) target_species genes remain after ortholog conversion.
#> 20,206 / 20,206 (100%) reference_species genes remain after ortholog conversion.
#> Retrieving all genes using: babelgene.
#> Retrieving all organisms available in babelgene.
#> Mapping species name: monkey
#> Common name mapping found for monkey
#> 1 organism identified from search: 9544
#> Using cached file: /github/home/.cache/R/orthogene/all_genes-9544-babelgene.csv.gz
#> Returning all 20,402 genes from monkey.
#> --
#> --
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from Gene.Symbol.
#> 20,402 genes extracted.
#> Converting monkey ==> Homo sapiens orthologs using: babelgene
#> Retrieving all organisms available in babelgene.
#> Mapping species name: monkey
#> Common name mapping found for monkey
#> 1 organism identified from search: Macaca mulatta
#> Retrieving all organisms available in babelgene.
#> Mapping species name: Homo sapiens
#> 1 organism identified from search: Homo sapiens
#> Retrieving all genes using: babelgene.
#> Retrieving all organisms available in babelgene.
#> Mapping species name: Macaca mulatta
#> 1 organism identified from search: 9544
#> Using cached file: /github/home/.cache/R/orthogene/all_genes-9544-babelgene.csv.gz
#> Checking for genes without orthologs in Homo sapiens.
#> Extracting genes from input_gene.
#> 20,402 genes extracted.
#> Extracting genes from ortholog_gene.
#> 20,402 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Dropping 1,920 genes that have multiple input_gene per ortholog_gene (many:1).
#> Dropping 1,357 genes that have multiple ortholog_gene per input_gene (1:many).
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 1,907 / 18,149 (11%)
#> Total genes remaining after convert_orthologs :
#> 16,242 / 18,149 (89%)
#> --
#>
#> =========== REPORT SUMMARY ===========
#> 16,242 / 18,149 (89.49%) target_species genes remain after ortholog conversion.
#> 16,242 / 20,206 (80.38%) reference_species genes remain after ortholog conversion.
#> Retrieving all genes using: babelgene.
#> Retrieving all organisms available in babelgene.
#> Mapping species name: mouse
#> Common name mapping found for mouse
#> 1 organism identified from search: 10090
#> Using cached file: /github/home/.cache/R/orthogene/all_genes-10090-babelgene.csv.gz
#> Returning all 29,651 genes from mouse.
#> --
#> --
#> Preparing gene_df.
#> data.table format detected.
#> Extracting genes from Gene.Symbol.
#> 29,651 genes extracted.
#> Converting mouse ==> Homo sapiens orthologs using: babelgene
#> Retrieving all organisms available in babelgene.
#> Mapping species name: mouse
#> Common name mapping found for mouse
#> 1 organism identified from search: Mus musculus
#> Retrieving all organisms available in babelgene.
#> Mapping species name: Homo sapiens
#> 1 organism identified from search: Homo sapiens
#> Retrieving all genes using: babelgene.
#> Retrieving all organisms available in babelgene.
#> Mapping species name: Mus musculus
#> 1 organism identified from search: 10090
#> Using cached file: /github/home/.cache/R/orthogene/all_genes-10090-babelgene.csv.gz
#> Checking for genes without orthologs in Homo sapiens.
#> Extracting genes from input_gene.
#> 29,651 genes extracted.
#> Extracting genes from ortholog_gene.
#> 29,651 genes extracted.
#> Checking for genes without 1:1 orthologs.
#> Dropping 9,436 genes that have multiple input_gene per ortholog_gene (many:1).
#> Dropping 10,451 genes that have multiple ortholog_gene per input_gene (1:many).
#> Filtering gene_df with gene_map
#> Adding input_gene col to gene_df.
#> Adding ortholog_gene col to gene_df.
#>
#> =========== REPORT SUMMARY ===========
#> Total genes dropped after convert_orthologs :
#> 4,848 / 20,075 (24%)
#> Total genes remaining after convert_orthologs :
#> 15,227 / 20,075 (76%)
#> --
#>
#> =========== REPORT SUMMARY ===========
#> 15,227 / 20,075 (75.85%) target_species genes remain after ortholog conversion.
#> 15,227 / 20,206 (75.36%) reference_species genes remain after ortholog conversion.
#> Loading required namespace: phytools
#> Loading required namespace: TreeTools
#> Registered S3 method overwritten by 'TreeTools':
#> method from
#> [.phyDat phangorn
#> Importing tree from: TimeTree2022
#> Importing cached tree.
#> Standardising tip labels.
#> Mapping 3 species from `species`.
#> Mapping 3 species from tree.
#> --
#> 0/3 (0%) tips dropped from tree due to inability to standardise names with `map_species`.
#> --
#> 0/3 (0%) tips dropped from tree according to overlap with selected `species`.
#> Gathering phylopic silhouettes.
#> Preparing data for 6 clades.
#> Warning: Each clade in `clades` must contain a vector of at least 1 species. Omitting clade: Invertebrates
#> 3 species remaining after metadata preparation.
#> Creating ggtree plot.
#> Found more than one class "phylo" in cache; using the first, from namespace 'tidytree'
#> Also defined by ‘TreeTools’
 #> Saving plot ==> /tmp/RtmpwPJAjL/file2e4264ce546f.ggtree.pdf
#> Saving plot ==> /tmp/RtmpwPJAjL/file2e4225496de3.ggtree.png
#> Saving plot ==> /tmp/RtmpwPJAjL/file2e4264ce546f.ggtree.pdf
#> Saving plot ==> /tmp/RtmpwPJAjL/file2e4225496de3.ggtree.png